Gene Expression and Splicing QTL Analysis of Blood Cells in African American Participants from the Jackson Heart Study
Our JHS-QTL is a queryable database for eQTLs and splicing QTLs (sQTLs) identified from 1,012 African Americans from the Jackson Heart Study [1], leveraging their PBMC RNA-sequencing data (~50 million reads per sample) and whole genome sequencing (WGS) data from the NHLBI Trans-Omics for Precision Medicine (TOPMed) program [2] (PMID: 33568819).
Our eQTL database contains a total of 4,524,238 variant-gene pairs significant at FDR 10% including 16,670 unique genes. Our sQTL database contains 7,419,368variant-gene-cluster pairs at FDR 10%, involving 9,872 unique genes.
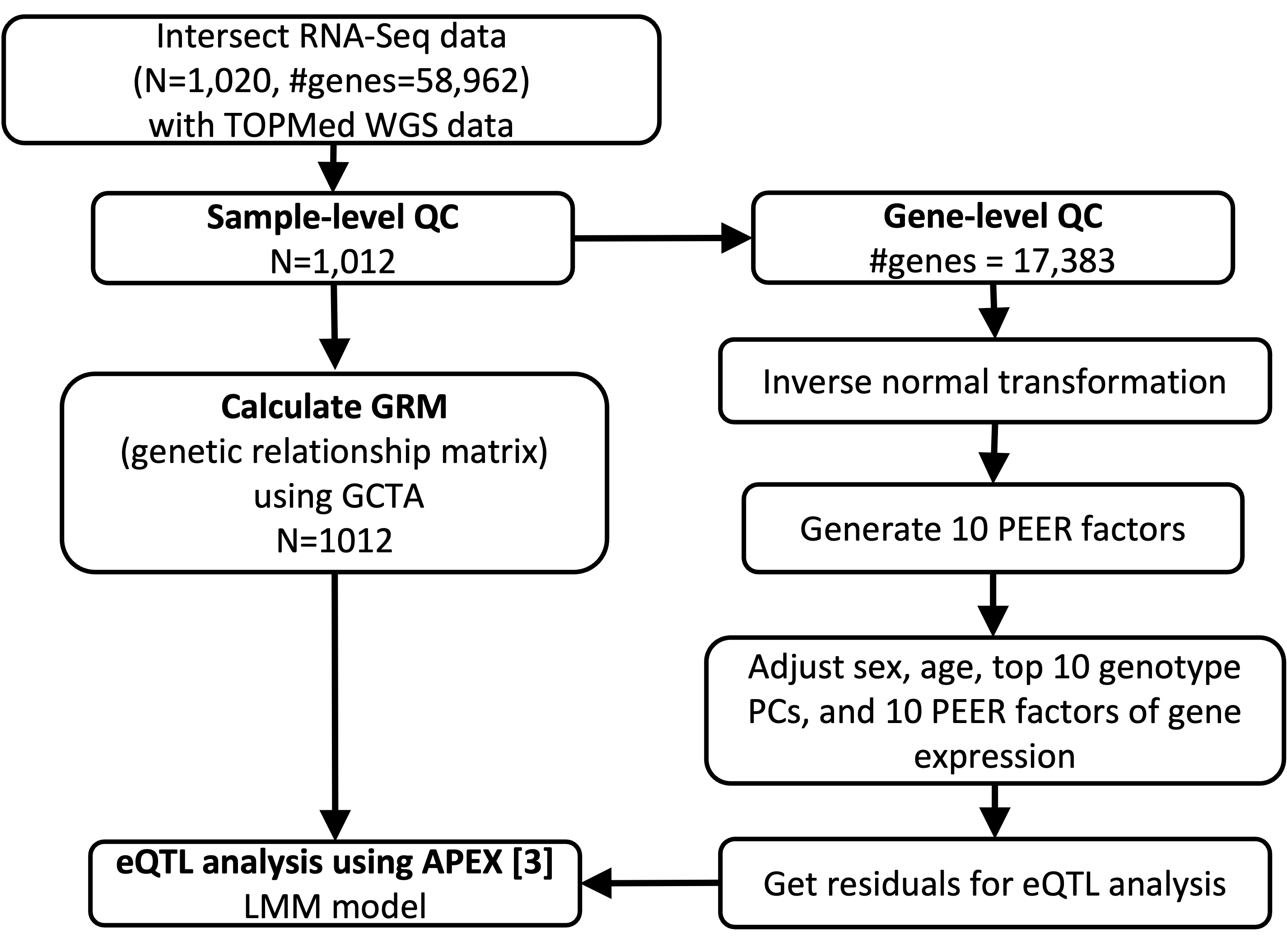
Quality Control and eQTL analysis pipeline
sQTL analysis pipeline
We followed the LeafCutter workflow [4] to identify sQTLs. Specifically, we quantified intron usage using LeafCutter, and then associated excision proportions of each intron cluster with genotypes of each variant in the +/-1Mb neighborhood to identify sQTLs using the APEX method [3]. Similar to eQTL analysis, we adjusted for age, sex, top 10 genotype PCs, and top 10 PCs from splicing quantification.
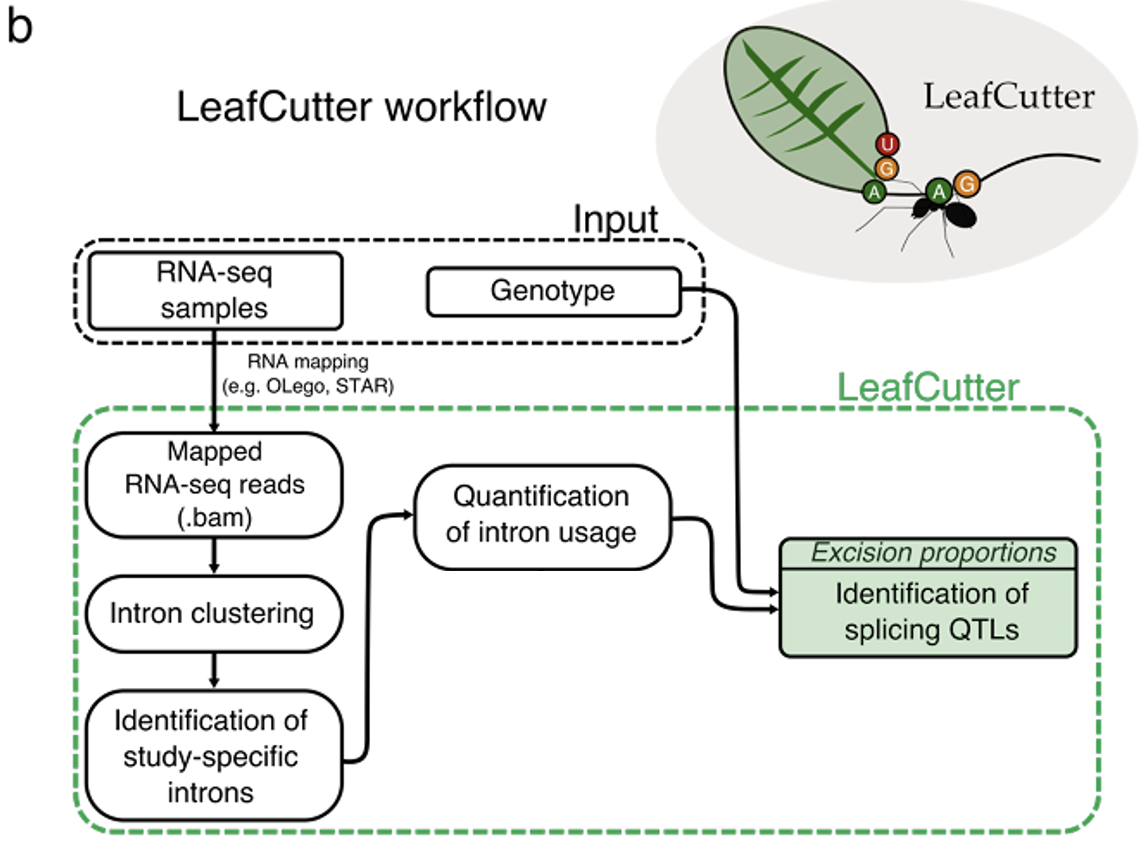
* Figure is taken from Figure 1 in Li et al 2018 Nat. Genet.
Citations
[1] Wilson, J. G., et al. (2005). "Study design for genetic analysis in the Jackson Heart Study." Ethn Dis 15(4 Suppl 6): S6-30-37.
[2].Taliun, Daniel, et al. "Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program." Nature 590.7845 (2021): 290-299.
[3] Quick, Corbin, et al. "A versatile toolkit for molecular QTL mapping and meta-analysis at scale." bioRxiv (2020).
[4] Li, Y.I., Knowles, D.A., Humphrey, J. et al. Annotation-free quantification of RNA splicing using LeafCutter. Nat Genet 50, 151-158 (2018). https://doi.org/10.1038/s41588-017-0004-9